Pre-clinical gene therapy protocols
1) Molybdenum Cofactor Deficiency
Molybdenum cofactor (MoCo) deficiency is an ultra-rare disease causing early neonatal death. MoCo is synthesized in the liver and functions as an essential co-factor for enzymes detoxifying sulphur components. During pregnancy maternal supply of MoCo prevents lesions, but very soon after birth highly toxic sulphur and xanthine deposits in the brain cause irreversible damages. MOCS1A/1B deficient mice were generated by Jochen Reiss (Dept. of Human Genetics, University Medicine Göttingen), and in a collaborative project we developed an AAV-based gene therapy protocol resulting in healthy animals after liver gene transfer (Kügler et al, Am J Hum Genet, 2007). Before such gene transfer very early after birth may become reality, several important questions must be addressed: due to the episomal nature of AAV vector genomes, they are heavily diluted by the massive cell divisions of hepatocytes during growth of the liver. Thus, will a single dose of vector applied into the neonatal liver result in sufficient MoCo supply for a human lifetime? Does this early application of AAV cause tolerance against the vector, allowing re-application at a later time without provoking immune reactions against the vector? And if tolerance against the therapeutic AAV is achieved, how will this impact on the natural infection with wild-type AAV that occurs in most humans during childhood?
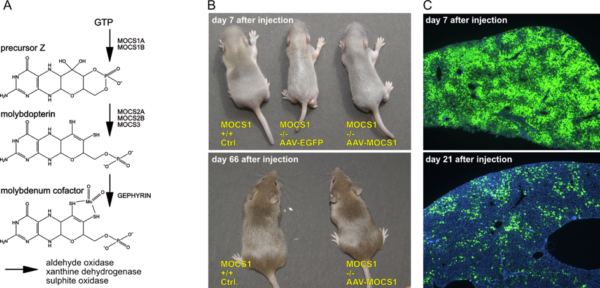
Molybdenum Cofactor (MoCo) gene therapy
- A) Biosynthesis of MoCo; the MOCS1A/1B genes are deficient in most patients.
- B) Control and MOCS1 -/- mice at 7 and 66 days after application of AAV-EGFP or AAV-MOCS1. AAV-EGFP injected animals die at 8-10 days after birth, while AAV-MOCS1 treated animals grow up with a normal phenotype.
- C) Dilution of AAV-vector genomes after intrahepatic injection as visualized by EGFP expression; DAPI nuclear counterstain in blue.