Vector tech and general applications
We develop and use AAV viral vectors for a wide variety of applications, i.e. for expression of Parkinson´s disease related proteins and genetically encoded sensors (e.g. for quantification of levels of calcium, ATP, ROS, cAMP etc.), both in cultured neurons and in the rodent brain. Expression of proteins or regulatory RNAs can be targeted to specific cell types or to specific sub-cellular organelles. The AAV toolkit offers extraordinary efficacy, safety and flexibility, in basic science but also in development of gene therapy.
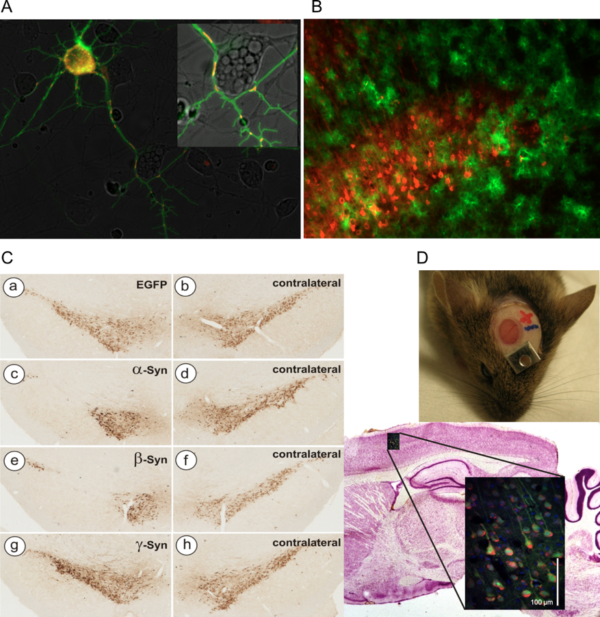
Examples of AAV vector applications routinely used in the lab
- A) Concomitant expression of cytoplasmic Ca2+ sensor GCaMP3.5 (green) and mitochondrial Ca2+ sensor RCaMP1e (red) in cultured neurons. Both sensors expressed by AAV-6 vectors under hSyn promoter.
- B) Mutually exclusive transduction of neurons (AAV-6-hSyn-Cherry, red) and astrocytes (AAV-5-GFAP-EGFP) in mouse cortical layer V.
- C) Neurodegeneration of nigral dopaminergic neurons after transduction with AAV-2 vectors expressing synuclein proteins (TH immunohistochemistry).
- D) Imaging of living neurons in mouse brain by 2-photon microscopy through a cranial window.
Pre-clinical gene therapy protocols
1) Molybdenum Cofactor Deficiency
Molybdenum cofactor (MoCo) deficiency is an ultra-rare disease causing early neonatal death. MoCo is synthesized in the liver and functions as an essential co-factor for enzymes detoxifying sulphur components. During pregnancy maternal supply of MoCo prevents lesions, but very soon after birth highly toxic sulphur and xanthine deposits in the brain cause irreversible damages. MOCS1A/1B deficient mice were generated by Jochen Reiss (Dept. of Human Genetics, University Medicine Göttingen), and in a collaborative project we developed an AAV-based gene therapy protocol resulting in healthy animals after liver gene transfer (Kügler et al, Am J Hum Genet, 2007). Before such gene transfer very early after birth may become reality, several important questions must be addressed: due to the episomal nature of AAV vector genomes, they are heavily diluted by the massive cell divisions of hepatocytes during growth of the liver. Thus, will a single dose of vector applied into the neonatal liver result in sufficient MoCo supply for a human lifetime? Does this early application of AAV cause tolerance against the vector, allowing re-application at a later time without provoking immune reactions against the vector? And if tolerance against the therapeutic AAV is achieved, how will this impact on the natural infection with wild-type AAV that occurs in most humans during childhood?
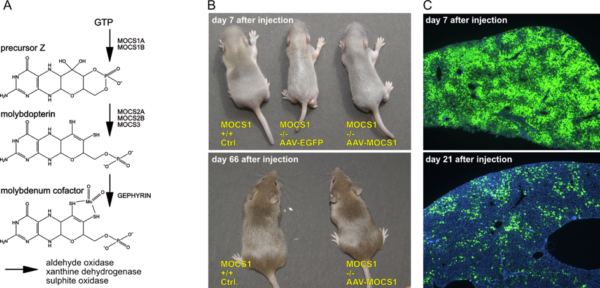
Molybdenum Cofactor (MoCo) gene therapy
- A) Biosynthesis of MoCo; the MOCS1A/1B genes are deficient in most patients.
- B) Control and MOCS1 -/- mice at 7 and 66 days after application of AAV-EGFP or AAV-MOCS1. AAV-EGFP injected animals die at 8-10 days after birth, while AAV-MOCS1 treated animals grow up with a normal phenotype.
- C) Dilution of AAV-vector genomes after intrahepatic injection as visualized by EGFP expression; DAPI nuclear counterstain in blue.
2) Pharmacological control of therapeutic transgene expression
Major neurodegenerative disorders like Alzheimer´s or Parkinson´s disease are characterized by a multifaceted etiology and disease progression. Thus, it is unlikely that a single target will be identified, modification of which might halt disease progression or even revert symptoms in the majority of idiopathic patients. To circumvent this issue, neurotrophic factors (NTFs), that are able to stimulate multiple survival-promoting pathways are considered a valuable option to prevent further loss of neurons and even to restore neuronal functionality. However, the enormous potency of these molecules is a solid basis for side effects as well. As NTFs cannot cross the blood-brain-barrier, their expression within the CNS by means of gene therapy is currently considered the optimal route of delivery. As a drawback, gene therapy in its current layout is an irreversible process, meaning that in case of excess supply of NTFs, expression cannot be stopped, nor can the expression level be regulated according to individual patient´s needs.
In order to provide an alternative, we have developed and further optimized an AAV vector system that takes advantage of pharmacological control over transgene expression. The FDA-approved human drug Mifepriston (Mfp), a synthetic steroid, controls expression of the neurotrophic factor GDNF through the so-called “GeneSwitch”, enabling us to tightly control the level and duration of GDNF expression in the brain. The system has proven to work favorably in a rat model of Parkinson´s disease, providing robust recovery from motor impairments (Maddalena et al, Mol Ther Nucl Acids, 2013; Tereshchenko et al, Neurobiol Dis, 2014; Chen et al, Exp Neurol, 2018). While in the immune-privileged environment of the human brain it is unlikely that the short yeast-derived peptide component of the “GeneSwitch” provokes immunological consequences, we still consider it to be necessary to conduct immunological studies in non-human primates, which are underway to demonstrate safety. Prove of absence of peripheral immunity against the “GeneSwitch” would also enable use of this regulated gene therapy concept in organs outside the CNS.
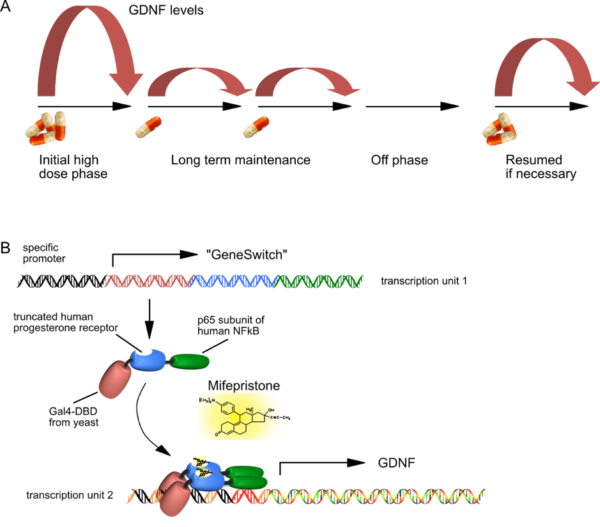
Pharmacological control of neurotrophic factor expression
- A) The principle of regulated GDNF expression is shown to demonstrate that different levels of Mifepristone can induce GDNF levels over two orders of magnitude.
- B) Basic principle of “GeneSwitch”-regulated expression. Both transcription units fit into a single AAV vector genome, and need a certain spatial configuration in order to allow for high-level but background-free GDNF expression. The “GeneSwitch” fusion protein consists primarily of human components, except for about 70 amino acids of the yeast DNA binding domain. Therefore, immunological studies in immunocompetent organs of non-human primates must now prove that this peptide will not be immunogenic in humans.
3) Dual-AAV for large transgenes: treatment of deafness
AAV are very small viruses, and thus can package only a quite limited transgene capacity of about 5000 base pairs, of which about 500-1000 bp are necessary for transcriptional control elements like promoter, enhancer and polyadenylation sites. This is unfortunate as several promising gene therapy candidates in sensory systems like retina or inner ear exceed this size by far. Due to their relatively small size, good accessibility and partial protection from immune responses towards the vectors, sensory organs are probably the best suited targets for successful gene therapy approaches in the near future. The Otoferlin protein is defective in a fairly large percentage of people suffering from hereditary hearing loss. Otoferlin is crucially involved into the auditory signal transmission process from inner hair cells to spiral ganglion neurons of the inner ear. As Otoferlin´s cDNA comprises about 6000 bp, it is not suitable to be packaged into a single AAV vector. In addition, we and other groups have demonstrated that vectors with enhanced transgene capacity such as lentiviral or adenoviral vectors are not suitable for inner hair cell transduction.
Thus, in a collaboration with Ellen Reisinger (Dept. of Otorhinolaryngology, University Medicine Göttingen) we exploited a dual AAV vector system in Otoferlin -/- mice, where the 5´-part of Otoferlin was incorporated into one AAV vector, and the 3´-part of Otoferlin was incorporated into a second AAA vector. Appropriately positioned splice sites allowed for reconstitution of full length Otoferlin cDNA in transduced inner hair cells, and enabled a partial but still impressive recovery of auditory function (Al-Moyed et al, EMBO Mol Med 2019). Confirmation in large animal´s ears will now have to prove that the vector application strategy is feasible in humans.
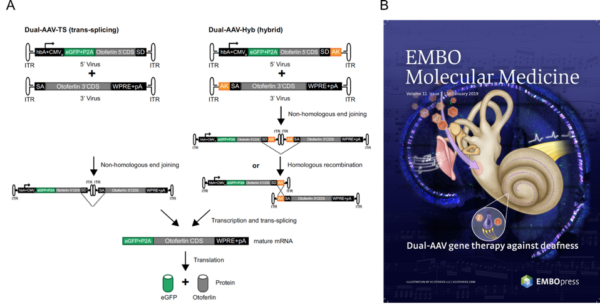
Dual AAV gene therapy in deaf Otoferlin -/- mice
- A) The two approaches used (trans-splicing genomes and hybrid genomes) to generate the full length Otoferlin construct are schematically depicted
- B) A nice representation of the study by EMBO´s artists.
Synuclein research 1: pathophysiological effects on mitochondrial integrity
The synuclein protein family consist of three closely related proteins, a-, b-, and g-Synuclein. They all show very little secondary or tertiary structure in solution, and are thus members of the ever-growing family of “unfolded” proteins. a-Synuclein can form protein aggregates under certain conditions, similar to other proteins linked to “aggregopathies” like Tau, Aß or Huntingtin. b-Synuclein was long thought to be an anti-aggregating counterpart to a-Synuclein, but we have recently demonstrated that it forms proteinase K-resistant aggregates in the rodent brain very similar to a-Synuclein (Taschenberger at al, Ann Neurol, 2013). a-Synuclein is intimately linked to the etiology of Parkinson´s disease (PD), as mutants and gene multiplications of a-Synuclein are directly causative for parkinsonian symptoms, and aggregations of a-Synuclein are found in all idiopathic PD patients. In order to better understand how a- but also ß-Synuclein inflict on neuronal physiology we use optical live imaging and biochemical tools in cultured primary neurons and in the rodent brain. An example of how we think pathophysiology is induced in mitochondria is illustrated below (deduced from Toloe at al, Front Mol Neurosci, 2018).
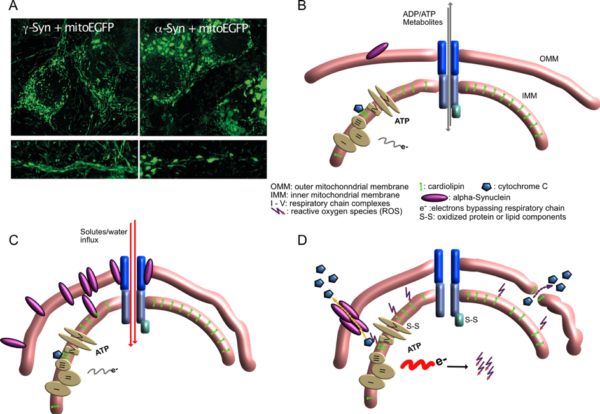
Mitochondrial lesions caused by a-synuclein
- A) Neurons overexpressing g-Synuclein show thin and elongated mitochondria in cytoplasm as well as in neurites (left panel), while mitochondria of a-Synuclein overexpressing neurons are swollen and condensed.
- B) Situation at the interface of inner and outer mitochondrial membrane under control conditions, when little a-Synuclein interacts with mitochondrial membranes.
- C) Under conditions of a-Synuclein overabundance it binds to mitochondrial membranes, causing enhanced membrane curvature and/or increased solute influx. Although mitochondria appear morphologically distorted at this stage, they are perfectly fine with respect to ATP production, ion handling capacities or ROS production.
- D) After longer-term interaction of a-Synuclein with mitochondrial membranes, ROS production increases, probably causing release of cytochrome C from its cardiolipin anchor (this part is speculative so far), resulting in even more loss of electrons from the respiratory chain and further oxidative damage. Finally, cytochrome C becomes released from mitochondria, either through a-Synuclein-generated pores or through damaged OMM, leading to induction of apoptosis-like neuronal cell death.
Synuclein research 2: pathophysiological effects on neuronal network activity
Neurons within the brain but also cultured primary neurons in a dish form interconnected networks which often show synchronized electrical activity without any external stimulation. We exploit this phenomenon to study the effects of the synucleins on neuronal activity and to elucidate the relative contribution of a-Synuclein´s interaction with ion channels, synaptic vesicle release and recycling or synaptic and neuritic structural integrity. This model allows to study pathophysiology induced by synucleins independent from neurodegeneration.
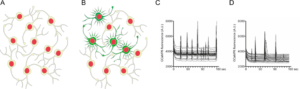
Neuronal network activity is affected by a-Synuclein
- A) Cultured primary neurons form dense network structures.
- B) A certain proportion of the neurons shows synchronized, non-stimulated electrical activity, which can be visualized by calcium imaging.
- C) Activity pattern of control neurons.
- D) a-Synuclein expressing neurons show significantly reduced network activity.